Le trône d'AlphaFold est menacé !
Nature publie un article : Le Biohub de Zuckerberg a sorti une bombe, publiant d'un coup 1,1 milliard de prédictions de structures protéiques, soit 800 millions de plus que la base de données d'AlphaFold.
Le modèle d'IA derrière, ESMFold2, serait plus performant qu'AlphaFold3 sur tous les plans.
Surtout, il est entièrement open-source, sans restriction d'usage commercial.

https://www.nature.com/articles/d41586-026-01686-3
La position dominante que Google DeepMind a patiemment construite durant des années dans le domaine de l'IA pour les protéines est ébranlée par un perturbateur open-source.
Le paysage de la course à l'IA pour les protéines pourrait être réécrit.
1,1 milliard de structures protéiques, servies d'un coup sur la table
Le 27 mai, le Biohub, une institution biomédicale créée par Mark Zuckerberg et sa femme, a officiellement lancé une base de données de structures protéiques nommée ESM Atlas.
1,1 milliard de structures protéiques prédites, accompagnées de 6,8 milliards d'informations de séquences protéiques.
La base de données d'AlphaFold a accumulé plus de 200 millions de prédictions de structures. ESM Atlas en ajoute d'emblée 800 millions.
Le modèle d'IA qui a généré ces prédictions s'appelle ESMFold2, développé sous la direction d'Alex Rives, responsable scientifique du Biohub.

Rives déclare :
Cette carte révèle le panorama complet de la biologie protéique, en particulier ses parties les plus inconnues.
Pourquoi la prédiction de la structure des protéines est-elle importante ?
Les protéines sont les pièces maîtresses du fonctionnement de la vie. Connaître leur forme permet de comprendre leur fonction, puis de concevoir de nouveaux médicaments et de vaincre des maladies.
C'est pour cela qu'AlphaFold a remporté le prix Nobel de chimie, un cas emblématique de l'IA transformant la science.
Maintenant, un nouveau modèle se dresse avec un jeu de données cinq fois plus grand.
En tant que modèle d'IA, où réside la force d'ESMFold2 ?
ESMFold2 emprunte une voie technique différente de celle d'AlphaFold.
Il est construit sur la base d'un « modèle de langage pour les protéines » publié en 2024. L'idée centrale s'inspire des pratiques du domaine du NLP (Traitement Automatique du Langage Naturel), traitant les séquences protéiques comme un « langage » à comprendre, en l'entraînant sur des dizaines de milliards de données protéiques, permettant au modèle d'apprendre à prédire directement la structure tridimensionnelle à partir de la séquence.
Les collègues en IA d'AlphaFold trouveront cela familier, c'est la même logique que celle des grands modèles de langage pour apprendre le langage humain.
L'étendue des données d'entraînement est une variable clé.
ESMFold2 intègre un grand nombre de données de protéines microbiennes provenant d'environnements comme le sol, les océans, etc., parties qui sont absentes de la base de données d'AlphaFold.
Une couverture plus large, le modèle a une vision plus complète du « monde protéique ».
L'équipe du Biohub affirme qu'ESMFold2 surpasse AlphaFold3 dans la prédiction des structures complexes des interactions entre protéines.
Mais ce qui est le plus convaincant, ce n'est pas les scores, c'est la validation expérimentale.
L'équipe a utilisé ESMFold2 pour concevoir de nouvelles protéines, les a synthétisées et testées en laboratoire, et une proportion élevée des conceptions ont fonctionné comme prévu.
La boucle « prédire » - « concevoir » - « valider » est bouclée, la valeur passe de l'article scientifique au monde réel.

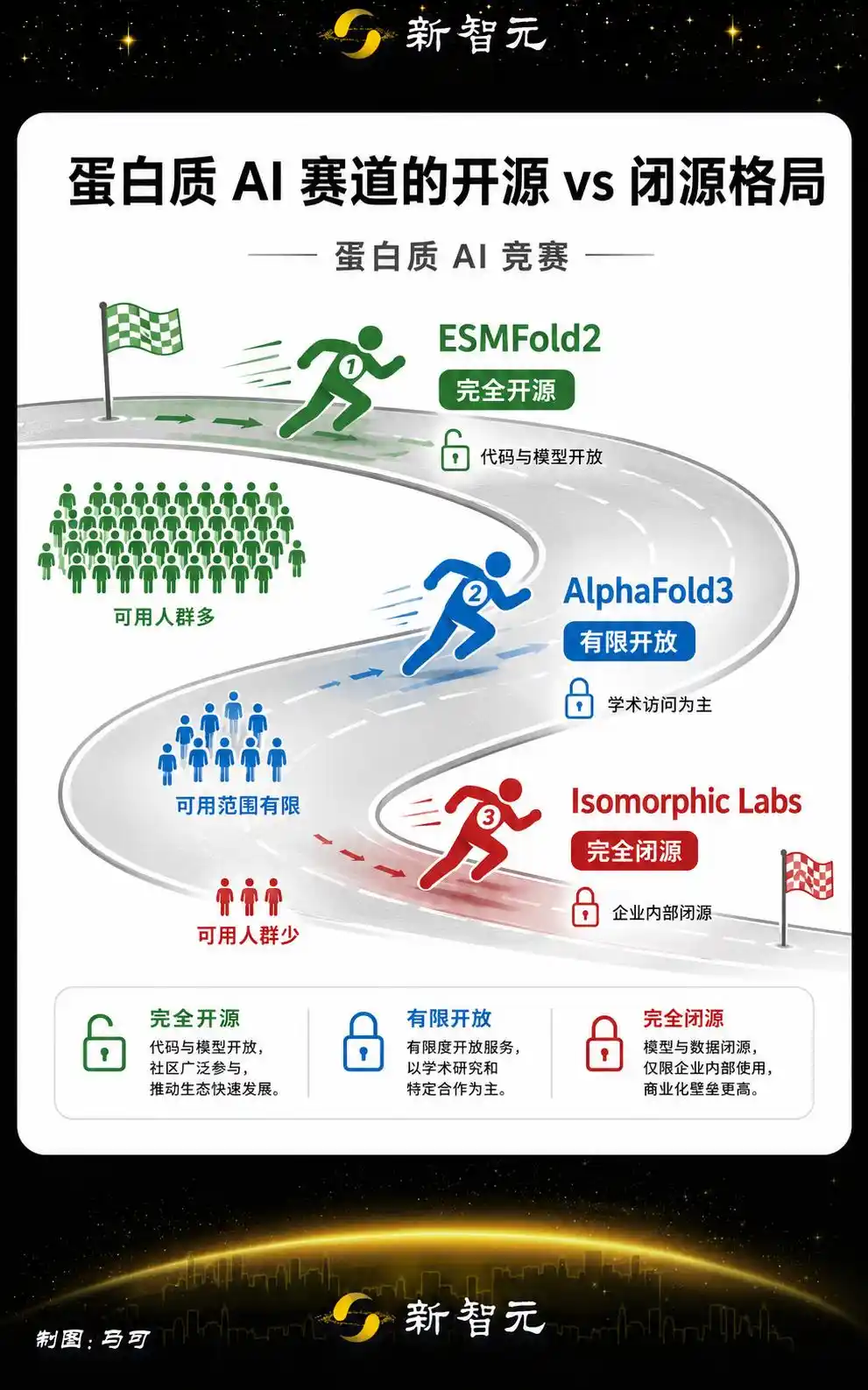
Entièrement open-source, voilà l'arme fatale
L'arme concurrentielle la plus tranchante d'ESMFold2 est d'être entièrement open-source et sans restriction commerciale.
La signification stratégique de ce choix est plus claire dans le contexte de l'ensemble de l'industrie de l'IA.
AlphaFold dispose d'une base de données ouverte, mais AlphaFold3 a initialement imposé des restrictions à l'usage commercial lors de sa publication.
Le modèle de prédiction des interactions protéiques lancé cette année par Isomorphic Labs, filiale de Google DeepMind, est totalement fermé.
Pour approfondir : Google publie « AlphaFold 4 », sans open-source ! Les performances écrasent la génération précédente.
Le biologiste computationnel du MIT, Ovchinnikov, souligne directement la valeur de l'open-source : « Je prévois que beaucoup seront très enthousiastes à l'idée d'essayer ESMFold2. »
L'effet de levier de l'IA open-source a déjà été pleinement démontré dans la course aux grands modèles de langage, la série Llama de Meta en est le meilleur exemple.
Un modèle open-source suffisamment puissant peut mobiliser la communauté mondiale pour l'itérer, l'appliquer, découvrir des utilisations auxquelles les développeurs originaux n'avaient même pas pensé.
La situation dans le domaine de l'IA pour les protéines est plus particulière, de nombreux laboratoires et institutions de recherche dans le monde ont un besoin urgent d'un outil de prédiction de structure gratuit et sans restrictions. Un modèle fermé, aussi puissant soit-il, ne touche qu'un nombre limité d'utilisateurs.
Le choix du Biohub d'être entièrement open-source s'inscrit dans la continuité de la stratégie de Meta avec les grands modèles de langage.
La stratégie de l'écosystème Zuckerberg dans le domaine de l'IA devient de plus en plus claire — utiliser l'open-source comme infrastructure, et l'écosystème comme rempart.
Les grands noms du domaine, sont-ils convaincus ?
Les réactions académiques sont positives, mais les réserves sont également claires.
Gemma Atkinson de l'Université de Lund en Suède qualifie l'ESM Atlas de « devrait être une ressource extraordinaire pour la biologie ».
Christine Orengo de l'University College London reconnaît sa valeur, mais souligne que les résultats prédits nécessitent une validation indépendante.

La question la plus pointue vient de Martin Steinegger de l'Université nationale de Séoul.

Il s'interroge sur la performance réelle d'ESMFold2 face à ces « nouvelles structures » très différentes des protéines connues.
Son équipe avait précédemment découvert que la première version d'ESMFold n'était pas excellente sur ce point. Cette question reste en suspens pour ESMFold2.
Ovchinnikov du MIT donne le jugement le plus lucide, estimant que l'ESM Atlas est mieux positionné comme complément à la base de données d'AlphaFold.

Il souligne également que le modèle fermé d'Isomorphic Labs ainsi que certains modèles open-source non directement comparés par le Biohub ont également obtenu des résultats similaires.
L'avance d'ESMFold2 pourrait ne pas être aussi importante que le suggère l'article.
Cette prudence reflète précisément que la compétition dans la course à l'IA pour les protéines est devenue féroce.
Open-source, fermé, académique, commercial, tous les types de modèles itèrent à une vitesse extrêmement rapide.
Le « plus fort » d'aujourd'hui pourrait être dépassé dans six mois. Ce rythme ressemble déjà beaucoup à la course aux armements dans le domaine des grands modèles de langage.
Quand l'IA commence à lire le code source de la vie
Autrefois, résoudre la structure tridimensionnelle d'une protéine pouvait nécessiter des mois, voire des années de travail en laboratoire.
AlphaFold a prouvé pour la première fois que l'IA pouvait le faire en quelques minutes.
Maintenant, ESMFold2 pousse l'échelle de prédiction au niveau du milliard, couvrant une grande quantité de protéines jamais résolues auparavant.
En poussant plus loin le raisonnement, lorsque l'IA pourra prédire avec précision toutes les structures protéiques, concevoir de nouvelles protéines fonctionnelles et les valider expérimentalement, alors la mise en œuvre d'une AGI (Intelligence Générale Artificielle) dans les sciences de la vie pourrait être plus proche que la plupart ne l'imaginent.
Si une ASI (Superintelligence Artificielle) arrive vraiment, la biologie ne sera plus pour elle une discipline à « étudier », mais un système pouvant être « ingénié ».
Concevoir la vie au niveau moléculaire, personnaliser les protéines à la demande, réécrire les règles de l'évolution.
Cela ressemble à de la science-fiction, mais des outils comme ESMFold2 transforment peu à peu la « science-fiction » en « problème d'ingénierie ».
Aujourd'hui, 1,1 milliard de structures protéiques sont étalées sur la table, accessibles gratuitement à tout scientifique dans le monde ayant une connexion internet.
Cela signifie que la capacité de l'IA à comprendre la vie a franchi une nouvelle étape.
Références : https://www.nature.com/articles/d41586-026-01686-3
Cet article provient du compte public WeChat « Nouvelle Intelligence Artificielle », auteur : Révélation ASI ; éditeur : Marco







