¡El trono de AlphaFold está en peligro!
Nature publica un artículo: Biohub, de Mark Zuckerberg, lanza una bomba definitiva, publicando de una sola vez 1.100 millones de predicciones de estructuras proteicas, 800 millones más que la base de datos de AlphaFold.
El modelo de IA detrás, ESMFold2, afirma superar completamente a AlphaFold3 en rendimiento.
Lo más crucial es que es completamente de código abierto, sin restricciones comerciales.

https://www.nature.com/articles/d41586-026-01686-3
La posición dominante en IA para proteínas que DeepMind de Google ha cultivado durante años, está siendo sacudida por un agitador de código abierto.
El panorama de la IA aplicada a las proteínas podría estar a punto de reescribirse.
1.100 millones de estructuras proteicas, servidas de una sola vez
El 27 de mayo, Biohub, la institución biomédica creada por los Zuckerberg, puso oficialmente en línea su base de datos de estructuras proteicas llamada ESM Atlas.
1.100 millones de estructuras proteicas predichas, junto con 6.800 millones de entradas de información de secuencias proteicas.
La base de datos de AlphaFold acumuló más de 200 millones de predicciones estructurales; ESM Atlas añade de golpe 800 millones más.
El modelo de IA que generó estas predicciones se llama ESMFold2, desarrollado por el equipo liderado por Alex Rives, responsable científico de Biohub.

Rives dice:
Este atlas revela la imagen completa de la biología proteica, especialmente de las partes más desconocidas.
¿Por qué es importante la predicción de la estructura proteica?
Las proteínas son los componentes centrales de la vida; conocer su forma permite comprender su función, diseñar nuevos fármacos y combatir enfermedades.
AlphaFold ganó el Premio Nobel de Química por esto, un caso emblemático de cómo la IA cambia la ciencia.
Ahora, un nuevo modelo aparece con un conjunto de datos 5 veces mayor.
Como modelo de IA, ¿en qué destaca ESMFold2?
ESMFold2 sigue un enfoque técnico diferente al de AlphaFold.
Se basa en un "modelo de lenguaje para proteínas" publicado en 2024. Su idea central se inspira en la PNL (Procesamiento del Lenguaje Natural), tratando las secuencias proteicas como un "lenguaje" para comprender, entrenando con decenas de miles de millones de datos proteicos, permitiendo al modelo aprender a predecir directamente la estructura tridimensional a partir de la secuencia.
Los colegas de IA de AlphaFold encontrarán esto familiar; es la misma lógica que usan los modelos de lenguaje grande para aprender el lenguaje humano.
La cobertura de los datos de entrenamiento es la variable clave.
ESMFold2 incluye una gran cantidad de datos de proteínas microbianas de entornos como el suelo y el océano, una brecha en la base de datos de AlphaFold.
Una cobertura más amplia significa un "mundo proteico" más completo para el modelo.
El equipo de Biohub afirma que ESMFold2 supera a AlphaFold3 en predecir estructuras complejas de interacciones entre proteínas.
Pero lo más convincente no son los benchmarks, sino la validación práctica.
El equipo usó ESMFold2 para diseñar proteínas completamente nuevas, las sintetizó y probó en el laboratorio, y un alto porcentaje de los diseños funcionó como se esperaba.
Pasar de la "predicción" al "diseño" y luego a la "validación" conecta una cadena que extiende el valor del artículo al mundo real.

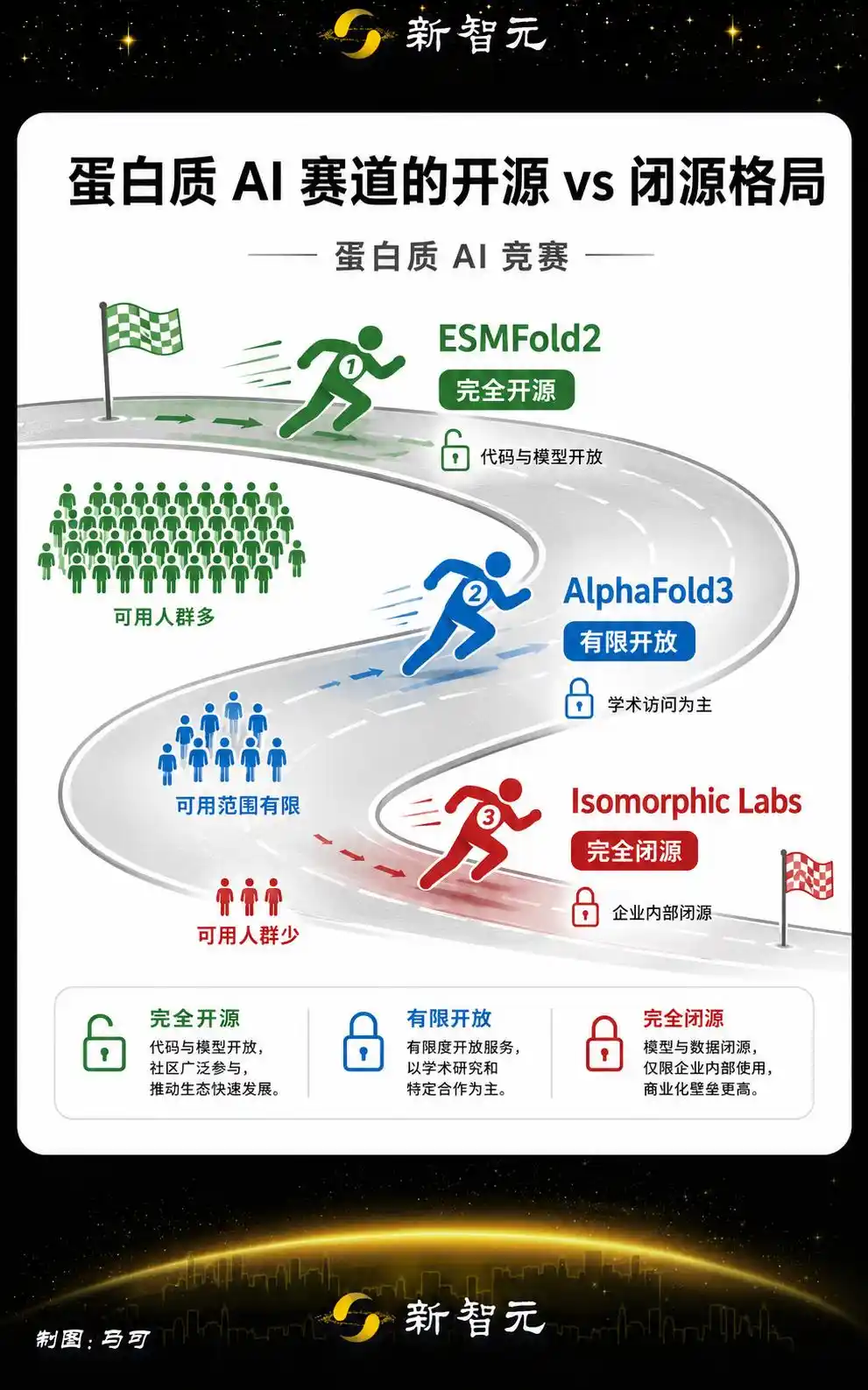
Completamente de código abierto, este es el arma más poderosa
El arma competitiva más afilada de ESMFold2 es ser completamente de código abierto y sin restricciones comerciales.
El significado estratégico de esta elección es más claro en el contexto de toda la industria de la IA.
AlphaFold tiene una base de datos abierta, pero AlphaFold3 inicialmente impuso restricciones al uso comercial.
Isomorphic Labs, de DeepMind de Google, lanzó este año un modelo de predicción de interacciones proteicas completamente cerrado.
Lectura relacionada: Google lanza "AlphaFold 4", ¡ya no es de código abierto! Rendimiento aplastante.
Ovchinnikov, biólogo computacional del MIT, señaló directamente el valor del código abierto: "Espero que mucha gente esté emocionada por probar ESMFold2."
El efecto palanca del código abierto en IA ya se ha demostrado en el campo de los LLM, y la serie Llama de Meta es el mejor ejemplo.
Un modelo de código abierto lo suficientemente potente puede movilizar a la comunidad global para iterar, aplicar y descubrir usos que ni siquiera los desarrolladores originales imaginaron.
La situación en el campo de la IA para proteínas es más especial: hay un gran número de laboratorios e instituciones de investigación en todo el mundo que necesitan urgentemente una herramienta de predicción estructural gratuita y sin restricciones. Por muy potente que sea un modelo cerrado, su alcance de usuarios es limitado.
La elección de Biohub de adoptar el código abierto completo sigue la misma estrategia que Meta en los LLM.
La estrategia del entorno de Zuckerberg en IA es cada vez más clara: usar el código abierto como infraestructura, y el ecosistema como barrera defensiva.
¿Los expertos del sector lo aceptan?
La respuesta académica es positiva, pero también hay reservas claras.
Gemma Atkinson, de la Universidad de Lund (Suecia), calificó a ESM Atlas como "debería ser un recurso extraordinario para la biología".
Christine Orengo, del University College London, reconoció su valor, pero enfatizó que los resultados de las predicciones necesitan verificación independiente.

Una pregunta más incisiva provino de Martin Steinegger de la Universidad Nacional de Seúl.

Le preocupa cómo se desempeña ESMFold2 frente a "nuevas estructuras" que difieren mucho de las proteínas conocidas.
Su equipo descubrió anteriormente que la primera versión de ESMFold no era excelente en este aspecto. Esta pregunta sigue sin respuesta para ESMFold2.
Ovchinnikov del MIT ofreció el juicio más sobrio, sugiriendo que ESM Atlas es más adecuado como complemento a la base de datos de AlphaFold.

También señaló que el modelo cerrado de Isomorphic Labs y algunos modelos de código abierto que Biohub no comparó directamente también han logrado resultados similares.
La ventaja de ESMFold2 podría no ser tan grande como sugiere el artículo.
Esta prudencia refleja precisamente que la competencia en el campo de la IA para proteínas ya es feroz.
Código abierto, cerrado, académico, comercial; todos los modelos están iterando a una velocidad extremadamente rápida.
El "más potente" de hoy podría ser superado en seis meses. Este ritmo ya se parece mucho a la carrera armamentística de los LLM.
Cuando la IA comienza a leer el código fuente de la vida
En el pasado, determinar la estructura tridimensional de una proteína podía llevar meses o años de trabajo de laboratorio.
AlphaFold demostró por primera vez que la IA podía hacerlo en minutos.
Ahora, ESMFold2 lleva la escala de predicción a 1.100 millones, cubriendo muchas proteínas nunca antes resueltas.
Extrapolando este camino, cuando la IA pueda predecir con precisión todas las estructuras proteicas, diseñar proteínas funcionales completamente nuevas y validarlas experimentalmente, entonces la implementación de la AGI en ciencias de la vida podría estar más cerca de lo que la mayoría cree.
Si llega la ASI (Superinteligencia Artificial), la biología dejaría de ser una disciplina que "estudiar", sino un sistema que puede "ingenierizarse".
Diseñar la vida a nivel molecular, personalizar proteínas bajo demanda, reescribir las reglas de la evolución.
Esto suena a ciencia ficción, pero herramientas como ESMFold2 están convirtiendo paso a paso la "ciencia ficción" en un "problema de ingeniería".
Hoy, 1.100 millones de estructuras proteicas están sobre la mesa, disponibles gratuitamente para cualquier científico del mundo con conexión a Internet.
Esto significa que la capacidad de la IA para comprender la vida ha dado otro paso adelante.
Referencias: https://www.nature.com/articles/d41586-026-01686-3
Este artículo proviene del WeChat Official Account "新智元" (Nueva Era de la Inteligencia), autor: ASI启示录; editor: Marco







